
Финансовые рынки – это государственные институты, которые обеспечивают движение капиталов между участниками хозяйственно – экономической деятельности РЫНОЧНОЙ экономики.
К таким участникам можно отнести Государство, государственные и коммерческие компании, институциональных и частных инвесторов. Цель одних участников – получение заёмных денег для выполнения поставленных задач в реальном секторе экономики, страхование будущих результатов своей хозяйственной деятельности, а также обеспечение финансовой связи с мировой экономикой, цель других – приумножение/преумножение денег.
Финансовые рынки призваны исполнить поставленные цели разными участниками.
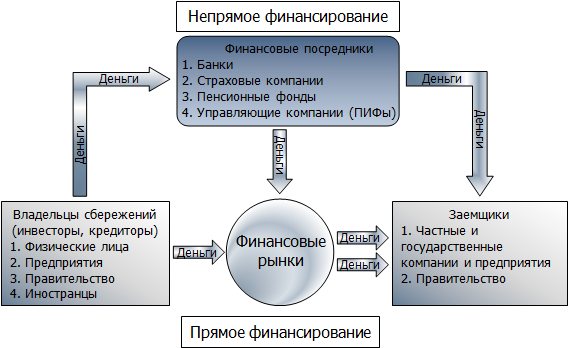
Рис. 1. Схема работы финансовых рынков.
На финансовых рынках свободные деньги направляются в финансовые активы либо напрямую от кредитора заёмщику в обмен на ценные бумаги, срочные обязательства и валюту, либо через посредников (непрямое финансирование) – деньги кредиторов аккумулируются в банках, страховых компаниях и различных фондах, а потом поступают заёмщикам.
Классификация финансовых рынков
По целям участников и выполняемым задачам финансовыми рынками последние можно разделить на:
- ➤ Валютный рынок
- ➤ Денежный рынок
- ➤ Долговой рынок
- ➤ Срочный рынок
- ➤ Товарный рынок
- ➤ Фондовый рынок
Краткое описание рынков
Мировая финансовая система представляет собой пирамиду, которую слагают финансовые рынки.
Фундаментом этой финансовой пирамиды является денежный рынок (банковский сектор). Своеобразная кровеносная система мировых финансов. Это рынок краткосрочных инвестиций. Совершая сделки деньги обмениваются на быстро реализуемые финансовые инструменты по рыночной стоимости. Фундаментальные показатели денежного рынка оценивают состояние ликвидности и уровня рисков в банковской системе. Денежный рынок во многом зависит от монитарной политики центральных банков и от политики Федеральной Резервной Системы США. Если ситуация на данном рынке ухудшается и банки начинают рассматривать риски контрагентов как высокие, то растёт стоимость фондирования. Чем выше ожидания негативных тенденций, тем дороже стоимость фондирования и выше плата за кредитные риски.
Середина финансовой иерархии есть долговой рынок. Или рынок капитала. Здесь обращаются долгосрочные финансовые инструменты. Чаще всего облигации. Размещая долговые бумаги эмитент привлекает средства на длительный срок, и может использовать их для разных целей. Например, финансирование бюджета или реализация инвестиционных программ. Участники рынка всегда дают оценку надёжности заёмщика, что выражается в доходности облигаций. Чем выше риски, тем выше стоимость заимствований. Однако, если центральные банки оказывают финансовое стимулирование экономики доходность может снижаться даже по облигациям не самых надёжных эмитентов. Таким образом, условия для заёмщика могут меняться от рыночной ситуации в лучшую сторону, независимо от состояния его экономики. Наличие развитого капитала в государстве считается главным признаком прогрессивной экономики. Чем больше развит рынок, тем больше развита экономика страны.
Завершает финансовую пирамиду фондовый рынок. Несмотря на свою известность и подробное обсуждение во всех СМИ данный рынок не является для финансовой системы ключевым. Но и у фондового рынка есть свои функции. Главной из которых является привлечение новых денег для бизнеса. Представители различных секторов экономики на определённом этапе своей хозяйственной деятельности проводят публичные размещения акций (IPO), фактически продавая часть своих компаний широкому кругу инвесторов. Таким образом, акции компаний попадают на биржу. В дальнейшем, оценивая перспективы развития компаний, участники фондового рынка покупают или продают акции этих компаний.
Взаимодействие и взаимозависимость этих трёх финансовых рынков происходит на базе валютного рынка FOREX. Он является основным и всеобъемлющим финансовым рынком. Потому что для совершения различных операций участникам финансовых рынков необходимо приобретать валюту той страны, в финансовые активы которой планируются вложения. Если инвестор хочет купить облигации Новой Зеландии, то он сначала должен купить на FOREX новозеландский доллар. Если долговые бумаги номинированы в евро или долларах, то необходимо сперва купить на валютном рынке эти валюты.
Валютный рынок необходим также в международной торговле. Для импорта и экспорта различных товаров на FOREX приобретают валюту расчёта.
Операции на данном финансовом рынке проходят непрерывно и в громадном объёме. Поэтому он является основополагающим для работы всей финансовой системы.





